Depósitos intracelulares
Cuando una célula en homeostasis es sometida a un estímulo patológico o a una mayor demanda fisiológica, tiene tres posibilidades:
Adaptarse a esa nueva demanda metabólica
Dañarse y presentar lesión (lesión reversible)
Sufrir un daño letal que la lleve a la muerte (lesión irreversible)
Podemos asumir como un signo de daño celularla presencia en la célula de contenidos anormalescon formación de gránulos o vacuolas
Origen de los contenidos anormales intracelulares:
Metabolismo celular
Espacio extracelular
Ambiente externo
Naturaleza química de los contenidos anormales:
Agua y electrolitos (TP 1) (Degeneración hidrópica, edema
celular)
Lípidos
Carbohidratos
Proteinas
Pigmentos (TP 4) (sustancias con coloración propia que la célula es incapaz de metabolizar)
Acumulación de lípidos
Triglicéridos --- Hígado, corazón, riñón --- Esteatosis
Colesterol --- Esteres
Fosfolípidos --- Figuras de mielina
Lipofucsina (pigmento)
Acumulación de triglicéridos (esteatosis)
Hígado, corazón, riñón
Proceso reversible
Fuentes de ácidos grasos:
Dieta (quilomicrones)
Movilización de ác. grasos desde tejido adiposo
Nueva síntesis a partir del acetato
Esteatosis hepática:
Aspecto macro:
Aumento del tamaño del órgano
Coloración amarillenta pálido, blanco
Friable, untuoso al corte
Esteatosis focal
Aspecto micro:
Degeneración celular con formación demicrovescículas hasta células de aspecto adipocitario (anillo de sello)
Motivos
Aumento del metabolismo lipídico por encima de la capacidad celular
Incapacidad celular de síntesis proteíca para el transporte de lípidos
Causas
Hipoxia (anemias, intoxicaciones) (disminución de la oxidación de ac. grasos)
Tóxicos, drogas (disminución de la síntesis protéica)
Aumento de la movilización y síntesis de ácidos garsos:
Malnutrición, dietas ricas en grasas, inanición
Obesidad
Hormonales
Metabólicas (preñez)
Acumulación de glucógeno:
Anomalía en el metabolismo de la glucosa (diabetes mellitus), el glucógeno (glucogenosis), hepatopatía inducida por esteroides, anoxia.
Microscópicamente: vacuolas claras en citoplasmaReacción de PAS +, tinción rosa a violeta del glucógeno
Acumulación de colesterol:
Aterosclerosis: músculo liso y macrófagos en íntima de grandes vasos
Macrófagos (células espumosas) en zonas de necrosis de tejido graso
Xantomas: subcutáneas, postraumáticas
Acumulación de proteínas:
Gotas hialinas: reabsorción en túbulos proximales renales
Enfermedades de depósito lisosomal:
Congénitas (mas de 30): esfingolipidosis, mucopolisacaridosis, lipidosis, glucoproteinosis, glucogenosis, lipofucsinosis ceroide
Adquiridas: drogas que alteran el pH lisosomal
Casuas
Actividad enzimática deficiente (hidrolasas específicas y coenzimas)
Incapacidad de transporte de material a través de la memebrana lisosomal
Depósitos extracelulares
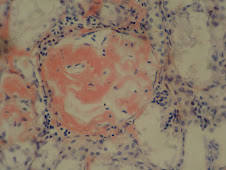
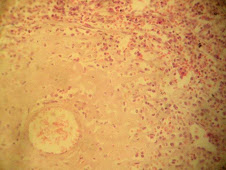
Amiloide
Proteínas fibrilares y glucoproteínas extracelulares, insolubles, patológicas, derivadas de precursores sanguíneos que se acumulan progresivamente
Microscópicamente: sustancia amorfa, hialina, eosinófila (H/E),rodea vasos sanguíneos y atrofia tejidos adyacentes por compresión
Refractario a muchos colorantes, Rojo Congo +, birefringencia verde con luz polarizada
Pobremente antigénico
Naturaleza física: filamentos de 7.5 a 10nm de diámetro, configuración en láminas cruzadas con plegamiento β
Naturaleza química: 90-95% proteínas fibrilares y 5-10% componente p (glucoproteína)
Dos tipos bien diferenciables AL y AA
Amiloidosis, clasificación:
Localizada
Formas cardíacas, cerebrales, endócrinas, asociadas con senilidad
Sistémica
Primaria
Asociadas a discracias plasmocitarias
Amiloide AL (cadenas livianas κ y λ) derivadas de células plasmáticas
Secundaria
Asociadas a procesos inflamatorios crónicos
Amiloide AA (amiloide asociado): proteína no inmunoglobulina derivada de un precursor proteíco sintetizado por el hígado
Ejemplos:
Mieloma --- Proliferación de plasmocitos productores de Ig --- Secreción de cadenas κ y λ --- Amiloide AL
Tuberculosis --- Acrivación de macrófagos --- Secreción deIL-1, IL-6 y TNF --- Estimulación hepática y secreción de SAA --- Amiloide AA
Cuando una célula en homeostasis es sometida a un estímulo patológico o a una mayor demanda fisiológica, tiene tres posibilidades:
Adaptarse a esa nueva demanda metabólica
Dañarse y presentar lesión (lesión reversible)
Sufrir un daño letal que la lleve a la muerte (lesión irreversible)
Podemos asumir como un signo de daño celularla presencia en la célula de contenidos anormalescon formación de gránulos o vacuolas
Origen de los contenidos anormales intracelulares:
Metabolismo celular
Espacio extracelular
Ambiente externo
Naturaleza química de los contenidos anormales:
Agua y electrolitos (TP 1) (Degeneración hidrópica, edema
celular)
Lípidos
Carbohidratos
Proteinas
Pigmentos (TP 4) (sustancias con coloración propia que la célula es incapaz de metabolizar)
Acumulación de lípidos
Triglicéridos --- Hígado, corazón, riñón --- Esteatosis
Colesterol --- Esteres
Fosfolípidos --- Figuras de mielina
Lipofucsina (pigmento)
Acumulación de triglicéridos (esteatosis)
Hígado, corazón, riñón
Proceso reversible
Fuentes de ácidos grasos:
Dieta (quilomicrones)
Movilización de ác. grasos desde tejido adiposo
Nueva síntesis a partir del acetato
Esteatosis hepática:
Aspecto macro:
Aumento del tamaño del órgano
Coloración amarillenta pálido, blanco
Friable, untuoso al corte
Esteatosis focal
Aspecto micro:
Degeneración celular con formación demicrovescículas hasta células de aspecto adipocitario (anillo de sello)
Motivos
Aumento del metabolismo lipídico por encima de la capacidad celular
Incapacidad celular de síntesis proteíca para el transporte de lípidos
Causas
Hipoxia (anemias, intoxicaciones) (disminución de la oxidación de ac. grasos)
Tóxicos, drogas (disminución de la síntesis protéica)
Aumento de la movilización y síntesis de ácidos garsos:
Malnutrición, dietas ricas en grasas, inanición
Obesidad
Hormonales
Metabólicas (preñez)
Acumulación de glucógeno:
Anomalía en el metabolismo de la glucosa (diabetes mellitus), el glucógeno (glucogenosis), hepatopatía inducida por esteroides, anoxia.
Microscópicamente: vacuolas claras en citoplasmaReacción de PAS +, tinción rosa a violeta del glucógeno
Acumulación de colesterol:
Aterosclerosis: músculo liso y macrófagos en íntima de grandes vasos
Macrófagos (células espumosas) en zonas de necrosis de tejido graso
Xantomas: subcutáneas, postraumáticas
Acumulación de proteínas:
Gotas hialinas: reabsorción en túbulos proximales renales
Enfermedades de depósito lisosomal:
Congénitas (mas de 30): esfingolipidosis, mucopolisacaridosis, lipidosis, glucoproteinosis, glucogenosis, lipofucsinosis ceroide
Adquiridas: drogas que alteran el pH lisosomal
Casuas
Actividad enzimática deficiente (hidrolasas específicas y coenzimas)
Incapacidad de transporte de material a través de la memebrana lisosomal
Depósitos extracelulares
Amiloide
Proteínas fibrilares y glucoproteínas extracelulares, insolubles, patológicas, derivadas de precursores sanguíneos que se acumulan progresivamente
Microscópicamente: sustancia amorfa, hialina, eosinófila (H/E),rodea vasos sanguíneos y atrofia tejidos adyacentes por compresión
Refractario a muchos colorantes, Rojo Congo +, birefringencia verde con luz polarizada
Pobremente antigénico
Naturaleza física: filamentos de 7.5 a 10nm de diámetro, configuración en láminas cruzadas con plegamiento β
Naturaleza química: 90-95% proteínas fibrilares y 5-10% componente p (glucoproteína)
Dos tipos bien diferenciables AL y AA
Amiloidosis, clasificación:
Localizada
Formas cardíacas, cerebrales, endócrinas, asociadas con senilidad
Sistémica
Primaria
Asociadas a discracias plasmocitarias
Amiloide AL (cadenas livianas κ y λ) derivadas de células plasmáticas
Secundaria
Asociadas a procesos inflamatorios crónicos
Amiloide AA (amiloide asociado): proteína no inmunoglobulina derivada de un precursor proteíco sintetizado por el hígado
Ejemplos:
Mieloma --- Proliferación de plasmocitos productores de Ig --- Secreción de cadenas κ y λ --- Amiloide AL
Tuberculosis --- Acrivación de macrófagos --- Secreción deIL-1, IL-6 y TNF --- Estimulación hepática y secreción de SAA --- Amiloide AA